GROUPING OF LYMPHOCYTE INTERACTIONS BY PARATOPE HOTSPOTS - GLIPH version 2
- Description
- GLIPH [Glanville J., Huang H. et al. Nature. 2017 Jul 6;547(7661):94-98] and GLIPH2 [Huang H., Wang C., et al. Nat Biotechnol 2020 Apr 27; https://doi.org/10.1038/s41587-020-0505-4] cluster TCRs that are predicted to bind the same MHC-restricted peptide antigen. When multiple donors have contributed to the clusters, and HLA genotypes for those donors are available, GLIPH additionally can provide predictions of which HLA-allele is presenting the antigen. Typically the user will pass in a set of hundreds to thousands of TCR sequences. This dataset will be analyzed for very similar TCRs, or TCRs that share CDR3 motifs that appear enriched in this set relative to their expected frequencies in an unselected naive reference TCR set. GLIPH returns significant motif lists, significant TCR convergence groups, and for each group, a collection of scores for that group indicating enrichment for motif, V-gene, CDR3 length, shared HLA among contributors, and proliferation count. When HLA data is available, it predicts the likely HLA that the set of TCRs recognizes.
- Updates from Version 1
-
- In version 2, a cluster based on local motif and a cluster based on global similarity are listed as two clusters even if these two clusters share one or more identical CDR3 sequences; while in version 1, these two clusters would be merged as one
- For a cluster based on local similarity (motif-based), motifs' position within CDR3 are restricted within 3 amino acids
- For a cluster based on global similarity, member CDR3s need to be of the same length, and differ at the same position. Users can choose to allow all amino acids exchangeable or only those amino acids with positive score in BLOSUM-62 matrix
- Reference data set is not required to be 10 times larger than the target data set
- Motifs overlapping with NP-amino acids are weighted more significant, where NP-amino acids are those encoded partially or entirely by non-template nucleotides during VDJ recombination
- Fisher-exact test is introduced to assess confidence of clusters
- TCR input file
- The format of TCR input file is tab-delimited, expecting the following columns in this order: CDR3b Vb Jb CDR3a Subject:condition Frequency
- HLA input file
- The format of HLA input file is also tab-delimited, with each row beginning with the identity of a subject, and then two or more following column providing HLA identification. The number of total columns (HLA defined genotypes) is flexible. Note that HLA input file is optional.
- Reference
- Users could choose either CD4, CD8 or CD48 reference according to from which subset of T cells those input TCR sequences are from.
- all_aa_interchangeable
- Users could choose either YES where all amino acids are interchangeable or NO where only those amino acids with positive scores in BLOSUM-62 matrix are interchangeable when computing global similarity.
- Demo data
- Demo data and results can be found at the link demo
- Example output
- The result is a csv file with format as below.
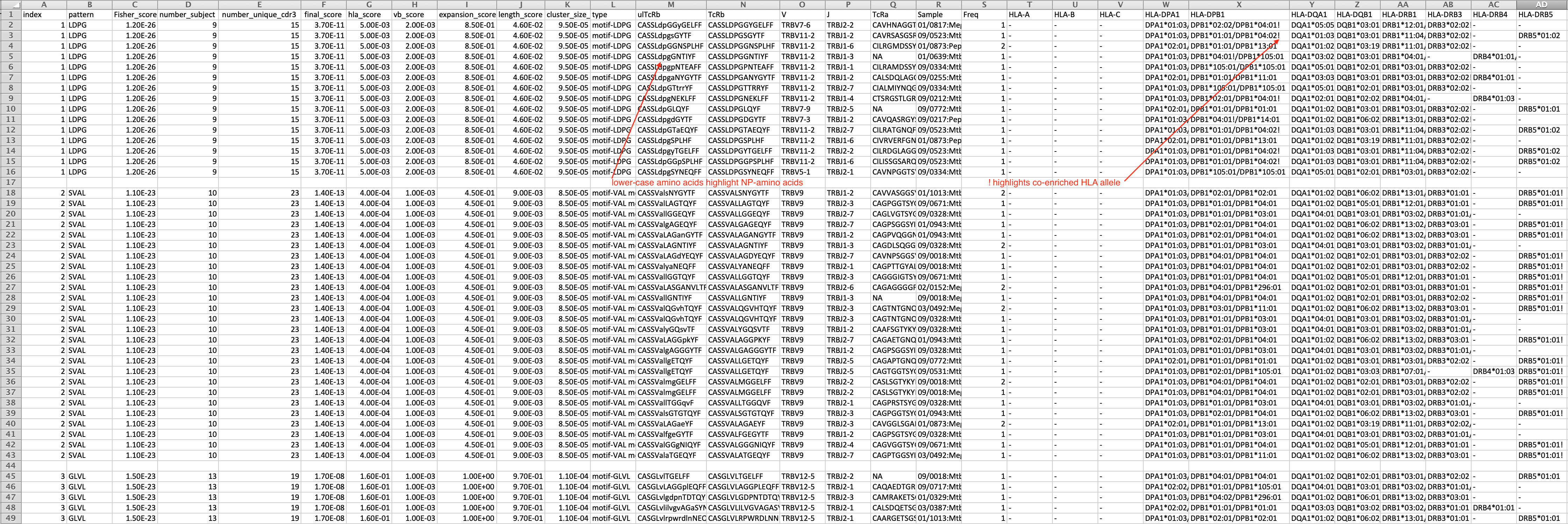
-
- index has no particular meaning
- pattern is the feature of the cluster
- Fisher_score estimates the bias of the pattern presenting in target data versus reference data using Fisher exact test
- number_subject is the number of subjects from which CDR3s are included in the cluster
- number_unique_cdr3: number of unique CDR3s in the cluster
- final_score, hla_score, vb_score, expansion_score, length_score, cluster_size_score are from GLIPH1
- type: global pattern contains '%', which indicates position allowing variants; local pattern starts with 'motif-'; and singlet likes global pattern without '%' symbol
- ulTcRb: amino acids encoded by codon overlapping with N-addition nucleotides are labeled as lower case
- Sample is the label of sample. If it is in the format "subject:condition", the first part is subject ID and the second part is the condition
- Freq is the count of cells in single cell data or the frequencies of CDR3s in bulk data.
- To use this web portal
- You need to register an account first; and then create a new project. Once a new project is created, the server will run GLIPH on the new project dataset automatically. You can checkout your results in a few minutes. It might take much longer for larger datasets. If you have questions to use this web portal, please feel free to write to chunlin@me.com or huang7@stanford.edu